the ylaboratory likes to ask 'why' questions
Naturally, the ‘y’ laboratory likes to ask ‘why’ questions: Why are some cells more vulnerable to Alzheimer’s disease? Why are some cancers more resistant to treatment than others? More broadly speaking, we seek to develop methods to tackle any interesting disease-related questions, whenever computation and data might help accelerate biological discoveries and enable potential improvements in quality of life: What can model organisms tell us about human disease? How can we develop better visualizations to aid interpretation and improve the efficiency of not only other computational biologists, but also bench scientists and clinicians?
With a solid computational foundation, we apply machine learning and quantitative reasoning to the following biological problems:
unraveling the complexity of neurological diseases
The complexity of the brain is highly challenging to model and understand. As a result, there are few safe and effective diagnostics and therapeutics for neurodegenerative diseases and psychiatric disorders. This is in no small part due to the fact that we have yet to develop a sufficient understanding of the molecular mechanisms that underlie these disorders. By modeling neurons and other specific cell types in the brain we are trying to unravel the complex dysfunctions occurring in Alzheimer's, Parkinson's, and other psychiatric disorders that have significant impacts on quality of life.

Spatial homology scores between mouse and human brains. Images depict the conservation of molecular identity of AD-vulnerable and resistent neuronal types between mouse and human brains.
selected related publications:
Disentangling associations between complex traits and cell types with seismic
Selective Neuronal Vulnerability in Alzheimer's Disease: A Network-Based Analysis
exploring the dynamic modulators of tumor biology
We develop computational models that integrate -omics data to uncover the mechanisms driving tumor behavior. Our analyses dissect the interplay of signaling pathways and cellular interactions while also considering factors such as microbial influences, splicing variations, and immune modulation, including prioritizing targets for engineered cell therapies. We are especially interested in modeling tumor heterogeneity and identifying promising avenues for therapeutic intervention.
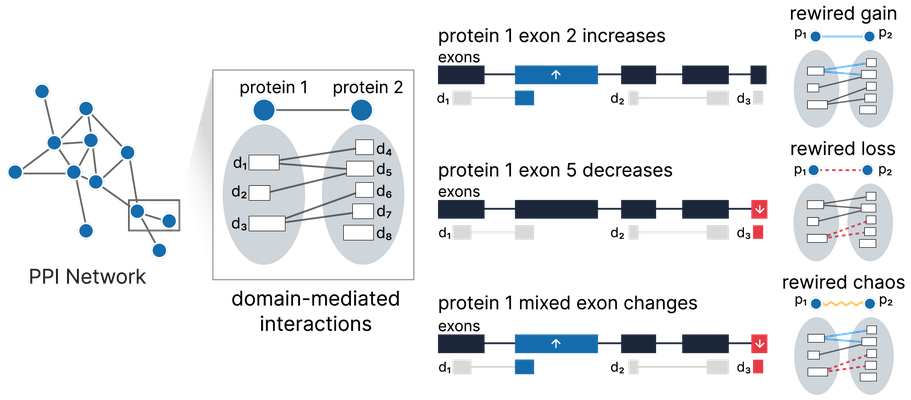
Overview of our Splitpea method for detecting PPI network rewiring due to alternative splicing. Splitpea combines prior knowledge in the form of protein-protein and
domain-domain interactions with splicing changes to provide a view of a rewired network for a given experimental context. A rewiring event occurs when exon changes affect an underlying domain-domain interaction. Here we show different scenarios that would result in one of the three possible rewiring events predicted by Splitpea.
selected related publications:
deciphering DNA methylation landscapes
We develop computational tools to map DNA methylation across tissues and bridge differences across experimental platforms to achieve comprehensive, genome-wide insights. Our work aims to capture how methylation patterns vary with tissue and cell type, and how external factors may influence these patterns to modulate gene regulation. This approach lays the groundwork for understanding the dynamic role of epigenetic regulation in cellular identity.
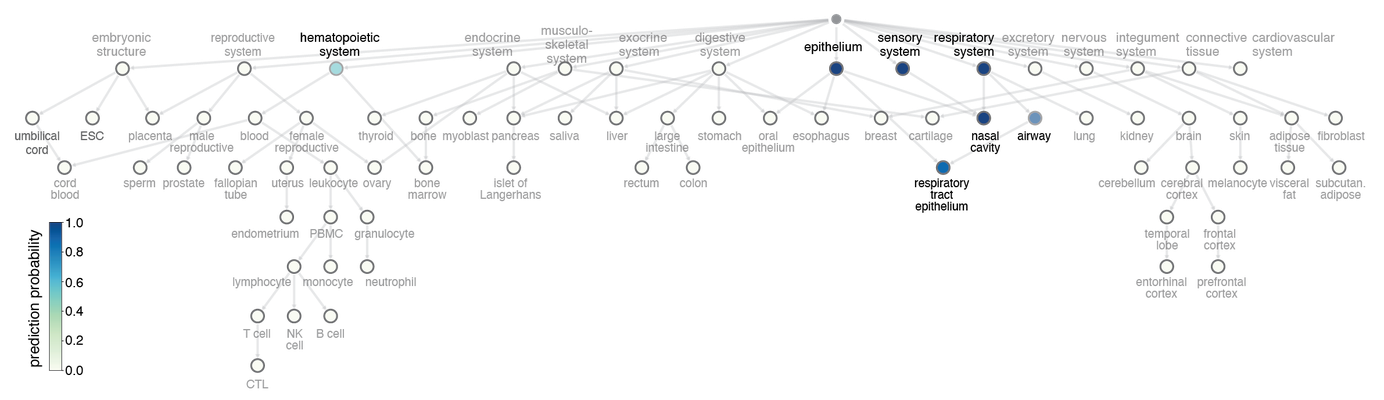
Label transfer prediction case study. Shown here is an example multi-label prediction for an epithelium of trachea sample (a label not seen in training) made with our ontology-aware DNA methylation classification method and visualized on a tissue / cell ontology. Nodes are colored by prediction probability.
selected related publications:
Ontology-aware DNA methylation classification with a curated atlas of human tissues and cell types
MONTE: Methylation-based Observation Normalization and Tumor purity Estimation
ARUNA: Slice-based self-supervised imputation for upscaling DNA methylation sequencing assays
extracting signals with interpretable embeddings
Modern biological data is vast and complex, making it challenging to uncover clear patterns. Our methods use advanced embedding techniques to compress high-dimensional data into simple, interpretable representations. By guiding these embeddings with known labels and creatively breaking down signals related to gene function, cellular states, and disease processes, we make data analysis more intuitive. This approach not only provides actionable insights for downstream prediction and cross-species analyses but also improves the integration of diverse datasets for better overall understanding.
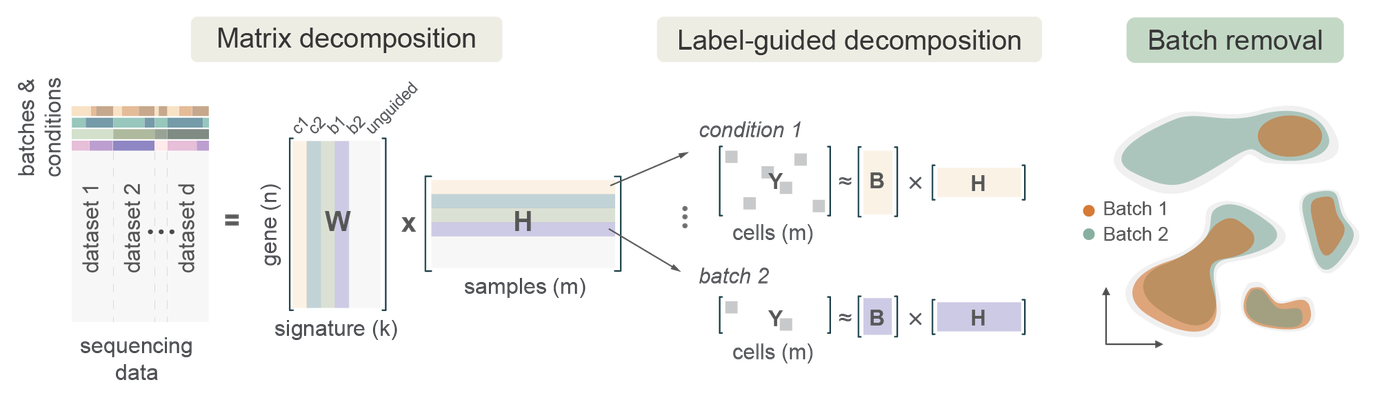
Overview of ALPINE. ALPINE is a semi-supervised non-negative matrix factorization method that can disentangle batch (e.g., dataset, kit, sequencing method) and biological variables of interest for extracting condition-specific signals from integrated datasets.
selected related publications:
Interpretable phenotype decoding from multi-condition sequencing data with ALPINE
A best-match approach for gene set analyses in embedding spaces
guiding discovery with networks and ontologies
Complex diseases arise from intricate interactions among genes, cells, and tissues. In our lab, we build robust network representations of gene relationships drawing on both functional and physical interaction data to design frameworks that incorporate biological structure and hierarchical information from curated ontologies. By constructing detailed models of tissue and cellular interactions, we uncover hidden disruptions that drive disease. Our integrative approaches can enable meaningful cross-species comparisons and be used to explore dynamic cellular states.
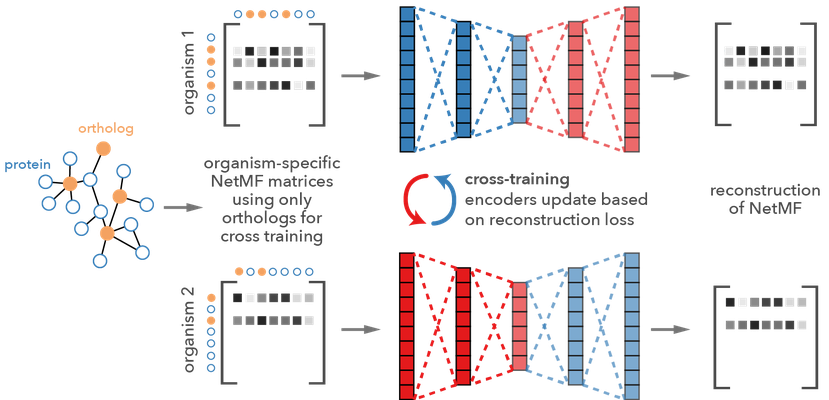
Brief overview of our method Embedding to Network Alignment (ETNA). The ETNA framework takes a pair of PPI network with a few ortholog anchor genes and aligns them using autoencoders.
selected related publications:
using NLP for reproducibility and knowledge extraction
To address reproducibility and transparency challenges, we harness natural language processing to automatically extract and annotate computational workflows from biomedical literature. By systematically charting the diverse pipelines employed in experimental and computational studies, we uncover common practices and potential pitfalls that may affect downstream analyses. Our approach not only streamlines the extraction of procedural knowledge but also evaluates the robustness of research findings, paving the way for more standardized and transparent practices in biomedical data science.
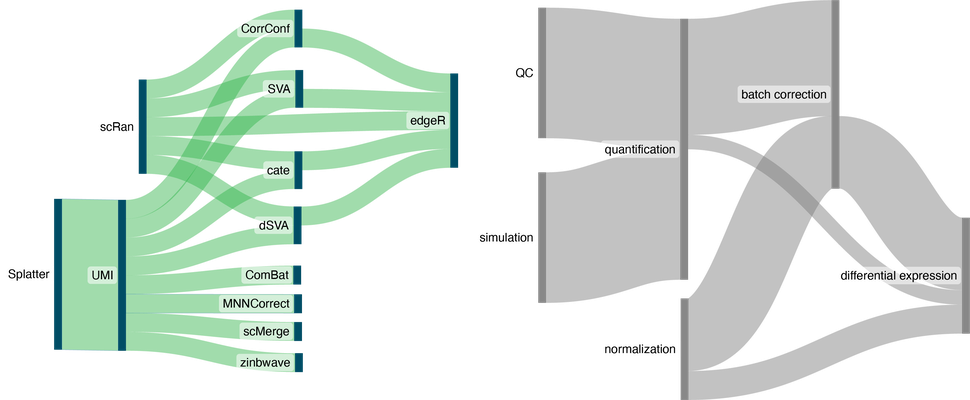
Sankey visualizations of tool and context workflows extracted from an example paper. Visualizations here reflect tools and the order in which they were applied (left) and the type of tools used (right).
selected related publications:
enabling complex data analyses with open-source tools
Methods and tools are best when they can be easily utilized by the greater community. We are strongly committed to developing tools and visualizations that help biologists and clinicians accelerate discovery. Dynamic, interactive systems combined with intuitive visualization can help unlock state of the art statistical and machine learning methods, enabling easy navigation of large, unwieldy, high-dimensional datasets, as well as models, even for those with no prior programming experience.